Quick Start#
Python scripts for these and other examples can be found here.
Diels Alder#
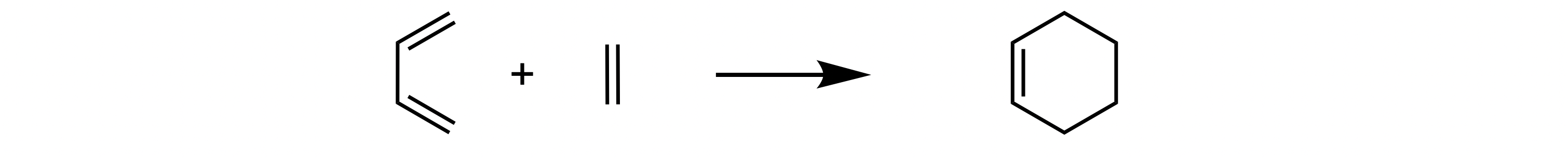
For the simple [4+2] Diels-Alder reaction between ethene and butadiene the reaction profile can be calculated in a couple of lines, where autodE identifies reactants and products from the reaction SMILES and executes using 8 CPU cores in 10 minutes or so using XTB and ORCA as the low and high level methods respectively.
>>> import autode as ade
>>> ade.Config.n_cores = 8
>>> rxn = ade.Reaction('C=CC=C.C=C>>C1=CCCCC1', name='DA')
>>> rxn.calculate_reaction_profile()
A directory (DA/) will be created where electronic structure calculations have been performed and an image of the reaction profile saved in the current working directory. See below:
SN2#
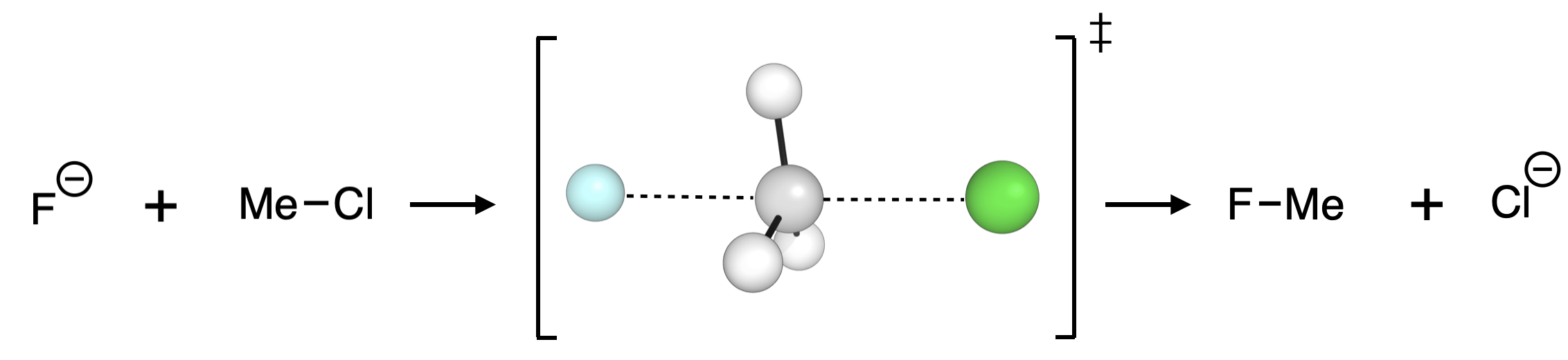
To generate a reaction profile for the SN2 reaction between fluoride and methyl chloride in water in more depth, we have the SMILES strings for the reactant and products generated from Chemdraw (by selecting a molecule → Edit → Copy As → SMILES):
Note
Fluoride: [F-]; MeCl: CCl; Chloride: [Cl-]; MeF: CF
Import autodE and set the number of processing cores that are available for this calculation:
>>> import autode as ade
>>> ade.Config.n_cores = 4
Initialise reactants and products from their respective SMILES strings giving a name to each:
>>> Fluoride = ade.Reactant(name='F-', smiles='[F-]')
>>> MeCl = ade.Reactant(name='CH3Cl', smiles='ClC')
>>> Chloride = ade.Product(name='Cl-', smiles='[Cl-]')
>>> MeF = ade.Product(name='CH3F', smiles='CF')
From reactants and products form a reaction in water and calculate the reaction profile:
>>> rxn = ade.Reaction(Fluoride, MeCl, Chloride, MeF, name='sn2', solvent_name='water')
>>> rxn.calculate_reaction_profile()
This function call will generate a plot something like:

as sn2_reaction_profile.png in the current working directory, where conformers of the reactant and products have been searched and the profile calculated at PBE0-D3BJ/def2-TZVP//PBE0-D3BJ/def2-SVP using an implicit water solvent. It should take around 10 minutes to complete.
Note
autodE has default DFT methods set for optimisation and single point calculations. Therefore, by default, structures are optimised at PBE0-D3BJ/def2-SVP and single points calculations performed at PBE0-D3BJ/def2-TZVP. To use other methods see the config page.