Reactions#
Reactions in autode are Reaction objects constructed from
either SMILES strings or Reactant and Product s. These are
elementary reactions, so the reactants should be linked to the products without
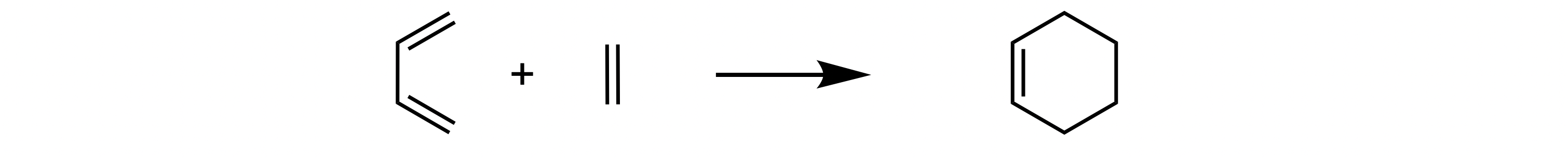
any intermediates. To initialise a reaction for: ethene + butadiene → cyclohexene:
>>> import autode as ade
>>>
>>> ethene = ade.Reactant(smiles='C=C')
>>> butadiene = ade.Reactant(smiles='C=CC=C')
>>> cyclohexene = ade.Product(smiles='C1=CCCCC1')
>>>
>>> rxn = ade.Reaction(ethene, butadiene, cyclohexene)
Reactions default to the gas phase and room temperature
>>> rxn.solvent is None
True
>>> rxn.temp # in K
298.15
Energy differences can be calculated for the overall reaction or to the
transition state (TS). If the energy of reactants and products has not been
calculated, then the energy differences are None
>>> rxn.delta('E') is None # ∆E_r
True
>>> rxn.delta('E‡') is None # ∆E‡
True
Calculating energies for the reactants and products allows for the reaction energy difference to be calculated
>>> for mol in (ethene, butadiene, cyclohexene):
... mol.optimise(method=ade.methods.XTB())
>>>
>>> rxn.delta('E').to('kcal mol-1')
Energy(-67.44178 kcal mol-1)
If a TS has not been located for the reaction then it is assumed to be barrierless and the barrier estimated from a diffusion limited process
>>> rxn.is_barrierless and rxn.delta('E‡').is_estimated
True
>>> rxn.delta('E‡').to('kcal mol-1')
Energy(4.35491 kcal mol-1)
To optimise the reactants and products then locate the transition state using 8 cores
>>> ade.Config.n_cores = 8
>>> rxn.optimise_reacs_prods()
>>> rxn.locate_transition_state()
>>>
>>> rxn.ts
TransitionState(TS_g1R2_X_ll_ad_2-3_4-5, n_atoms=16, charge=0, mult=1)
>>> # ∆E‡ is now no longer an estimate
>>> rxn.delta('E‡').to('kcal mol-1')
Energy(14.30068 kcal mol-1)
Identity reactions where reactants and products are identical are not, by default, supported in autode as the bond rearrangement of interest is not easily inferred. However, reaction profiles for identity reactions may be calculated by defining atom classes to distinguish otherwise identical atoms. For example
>>> rxn = ade.Reaction('[Br-:1].C[Br:2]>>C[Br:1].[Br-:2]', solvent_name='water')
>>> # bond rearrangement leading to products is well defined
>>> rxn.calculate_reaction_profile()
calculates the profile for the Br- + CH3Br -> BrCH3 + Br- SN2 reaction. An
atom.atom_class attribute is set when defined in the SMILES string. This
may be set directly, with the following two molecules being identical
>>> mol_a = ade.Molecule(smiles='[He:1]')
>>> mol_b = ade.Molecule(atoms=[ade.Atom('He', atom_class=1)])
>>> mol_a.atoms[0].atom_class == mol_b.atoms[0].atom_class == 1
True