Molecules#
Reactants and Products are Molecules and are initialised much like their Species parent, but have charge and multiplicity defaults (0, 1 respectively) and can be built from SMILES strings
>>> import autode as ade
>>> molecule = ade.Molecule(name='molecule')
>>> molecule.charge
0
>>> molecule.mult
1
or from 3D structures given as xyz files directly
>>> ch4 = ade.Molecule('methane.xyz')
>>> ch4.name
'methane'
Simple Example#
To generate a water Molecule from its SMILES string (‘O’), where hydrogen atoms are implied
>>> water = ade.Molecule(name='h2o', smiles='O')
>>> water.atoms
Atoms(n_atoms=3, [Atom(O, -0.001, 0.363, -0.000),
Atom(H, -0.825, -0.182, -0.000),
Atom(H, 0.826, -0.181, 0.000)])
Molecules also add a molecular graph attribute as a NetworkX
Graph and contain node (atoms) and edge (bonds) attributes
>>> water.graph
MolecularGraph(|E| = 2, |V| = 3)
>>> water.graph.nodes
NodeView((0, 1, 2))
>>> water.graph.edges
EdgeView([(0, 1), (0, 2)])
where in water there are three atoms {0, 1, 2} and two bonds. The 3D structure can be generated as a .xyz file for viewing in molecular visualisation software (Avogadro, Chimera, VMD, Mercury, Maestro etc.) with
>>> water.print_xyz_file()
where ‘h2o.xyz’ is generated in the current working directory.
Geometry Manipulation#
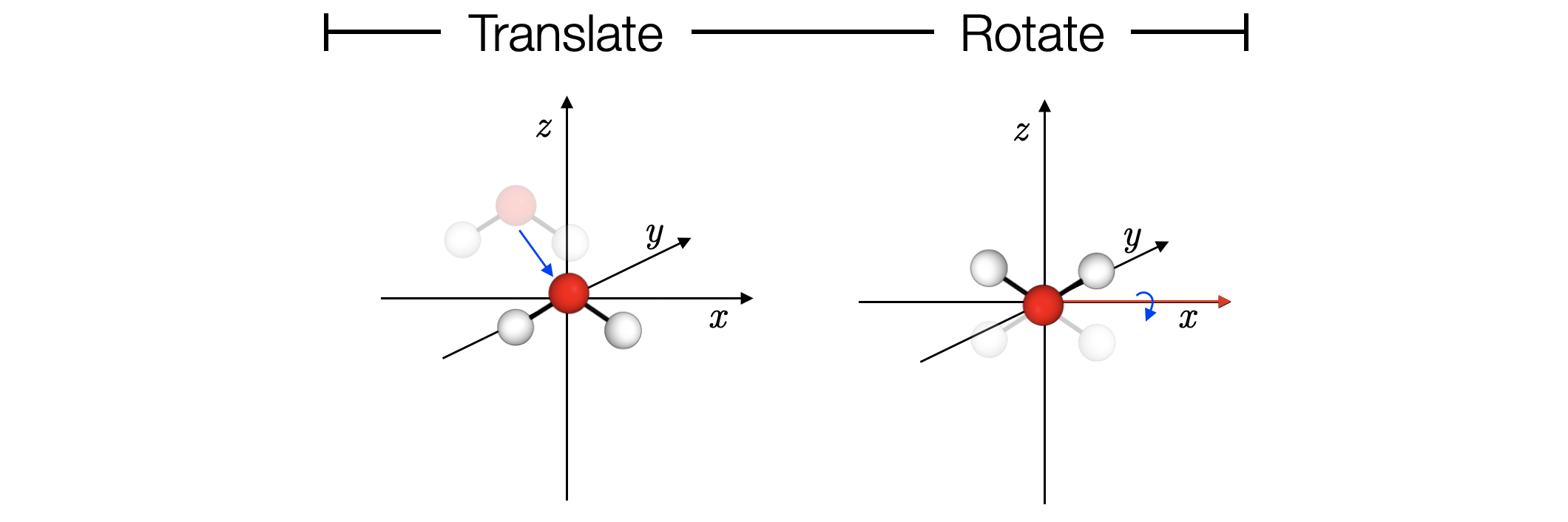
Whole molecules can be translated and rotated. For example, to translate the water molecule so the oxygen atom is centred at the origin
>>> water.coordinates
Coordinates([[-0.0011, 0.3631, -0. ],
[-0.825 , -0.1819, -0. ],
[ 0.8261, -0.1812, 0. ]])
>>> o_atom = water.atoms[0]
>>> water.translate(vec=-o_atom.coord)
>>> water.coordinates
Coordinates([[ 0. , 0. , 0. ],
[-0.8250, -0.1819, 0. ],
[ 0.8261, -0.1812, 0. ]])
then rotate around the x axis
>>> import numpy as np
>>> water.rotate(axis=[1.0, 0.0, 0.0], theta=np.pi)
>>> water.coordinates
Coordinates([[ 0. , 0. , 0. ],
[-0.8250, 0.1819, 0. ],
[ 0.8261, 0.1812, 0. ]])
Angles between atoms in a molecule can be also calculated
>>> water.angle(1, 0, 2)
Angle(1.9752 rad)
where atoms are indexed from 0, so the angle is θ(H-O-H). As with distances, explicit unit conversion is supported
>>> water.angle(1, 0, 2).to('deg')
Angle(113.17085 °)
Calculations#
autodE provides wrappers around common electronic structure theory packages (ORCA, XTB, NWChem, MOPAC, Gaussian09, Gaussian16, QChem) so geometries may be optimised and energies calculated.
For example, to optimise the geometry of a water molecule at the XTB level and then perform a single point energy evaluation with ORCA
>>> water.optimise(method=ade.methods.XTB())
>>> water.energy
Energy(-5.07054 Ha)
>>> water.single_point(method=ade.methods.ORCA())
>>> water.energy
Energy(-76.37766 Ha)
where the default single point method in ORCA is PBE0-D3BJ/def2-TZVP. Like with other values (distances, angles, dihedrals) converting to different units is as simple as
>>> water.energy.to('kcal')
Energy(-47927.6682 kcal mol-1)
water.energy returns the most recently evaluated energy at this geometry,
but the XTB energy is still saved in water.energies. Printing the energies
along with their associated methods
>>> for energy in water.energies:
... energy, energy.method_str
...
Energy(-5.07054 Ha) xtb
Energy(-76.37766 Ha) orca PBE0-D3BJ/def2-TZVP
Modifying the method is possible by setting keywords. To set the single point keywords for an instance of the ORCA wrapper
>>> orca = ade.methods.ORCA()
>>> orca.keywords.sp = ['PBE0', 'D3BJ', 'ma-def2-TZVP']
>>> water.single_point(method=orca)
>>> water.energy
Energy(-76.37938 Ha)
Keywords can also be passed as arguments to single_point, optimise
and calc_thermo. For example:
>>> water.single_point(method=ade.methods.ORCA(),
... keywords=['PBE0', 'D3BJ', 'ma-def2-TZVP'])
will do an identical calculation to the above example.
Alternatively, to set the keywords for every instance of ORCA created,
use ade.Config e.g.
>>> ade.Config.ORCA.keywords.sp = ['PBE0', 'D3BJ', 'ma-def2-TZVP']
>>> instance_1 = ade.methods.ORCA()
>>> instance_1.keywords.sp
SPKeywords(PBE0 D3BJ ma-def2-TZVP)
>>> instance_2 = ade.methods.ORCA()
>>> instance_2.keywords.sp
SPKeywords(PBE0 D3BJ ma-def2-TZVP)
Note
Structure optimisation resets the positions of the atoms to their optimised value.
Calculations can also be performed using electronic structure packages with implemented wrappers. For example, to calculate a single point energy for a hydrogen atom with all the currently implemented methods
>>> from autode.methods import MOPAC, XTB, QChem, NWChem, G09, G16, ORCA
>>>
>>> h = ade.Molecule(name='H', mult=2, atoms=[ade.Atom('H')])
>>>
>>> h.single_point(method=MOPAC())
>>> h.single_point(method=XTB())
>>> h.single_point(method=QChem())
>>> h.single_point(method=NWChem())
>>> h.single_point(method=G09())
>>> h.single_point(method=G16())
>>> h.single_point(method=ORCA())