Non-covalent Interaction Complexes#
autodE allows for the systematic search of a NCI complexes conformational space. For example, to find the lowest energy structure of the water trimer:
import autode as ade
xtb = ade.methods.XTB()
# Number of points on the surface of the sphere for each component
ade.Config.num_complex_sphere_points = 5 # N_s
# and the number of rotations to perform per point on the sphere
ade.Config.num_complex_random_rotations = 3 # N_r
# Total number of conformers ~(N_s × N_r)^(N-1) for N molecules => ~225
# Make a water molecule and optimise at the XTB level
water = ade.Molecule(name="water", smiles="O")
water.optimise(method=xtb)
# Make the NCI complex and find the lowest energy structure
trimer = ade.NCIComplex(water, water, water, name="water_trimer")
trimer.find_lowest_energy_conformer(lmethod=xtb)
trimer.print_xyz_file()
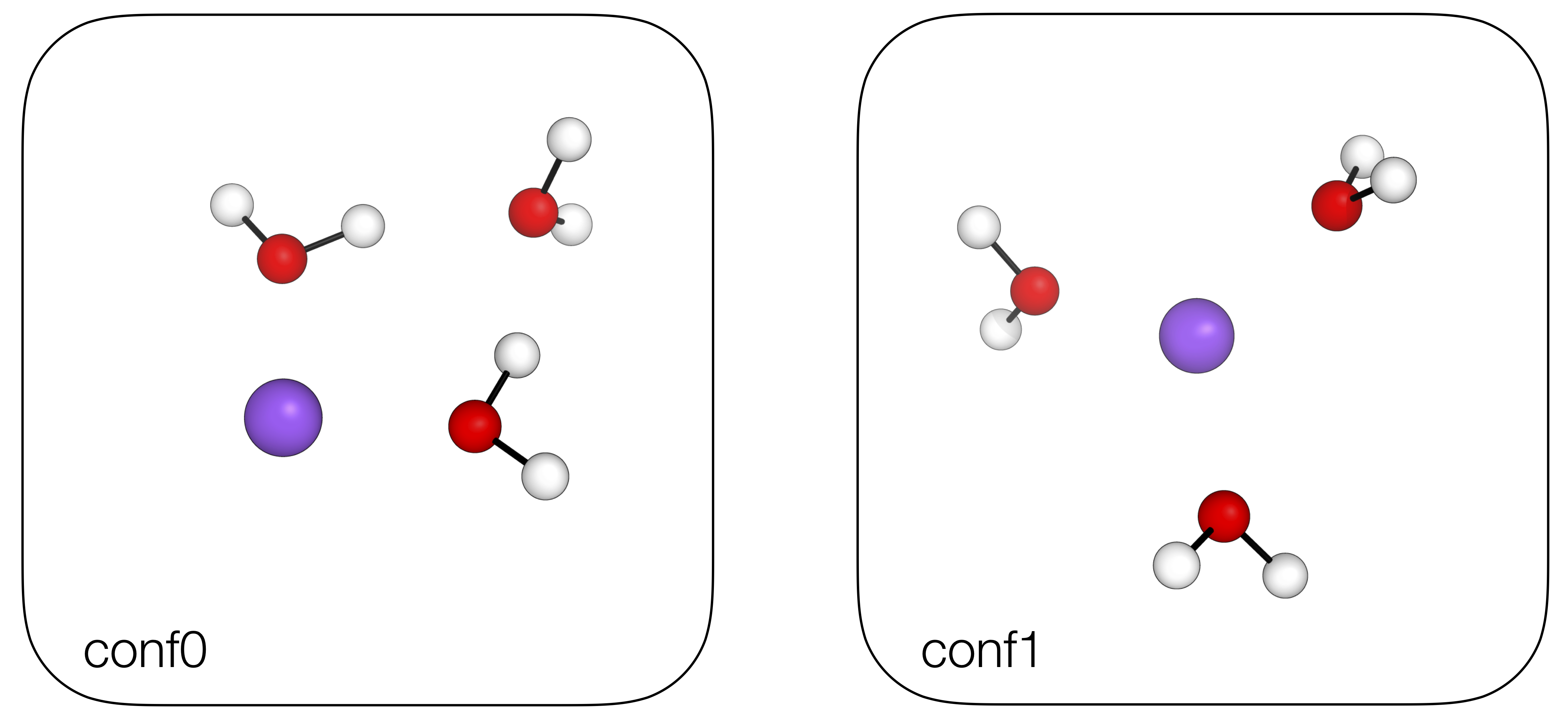
Out (visualised)
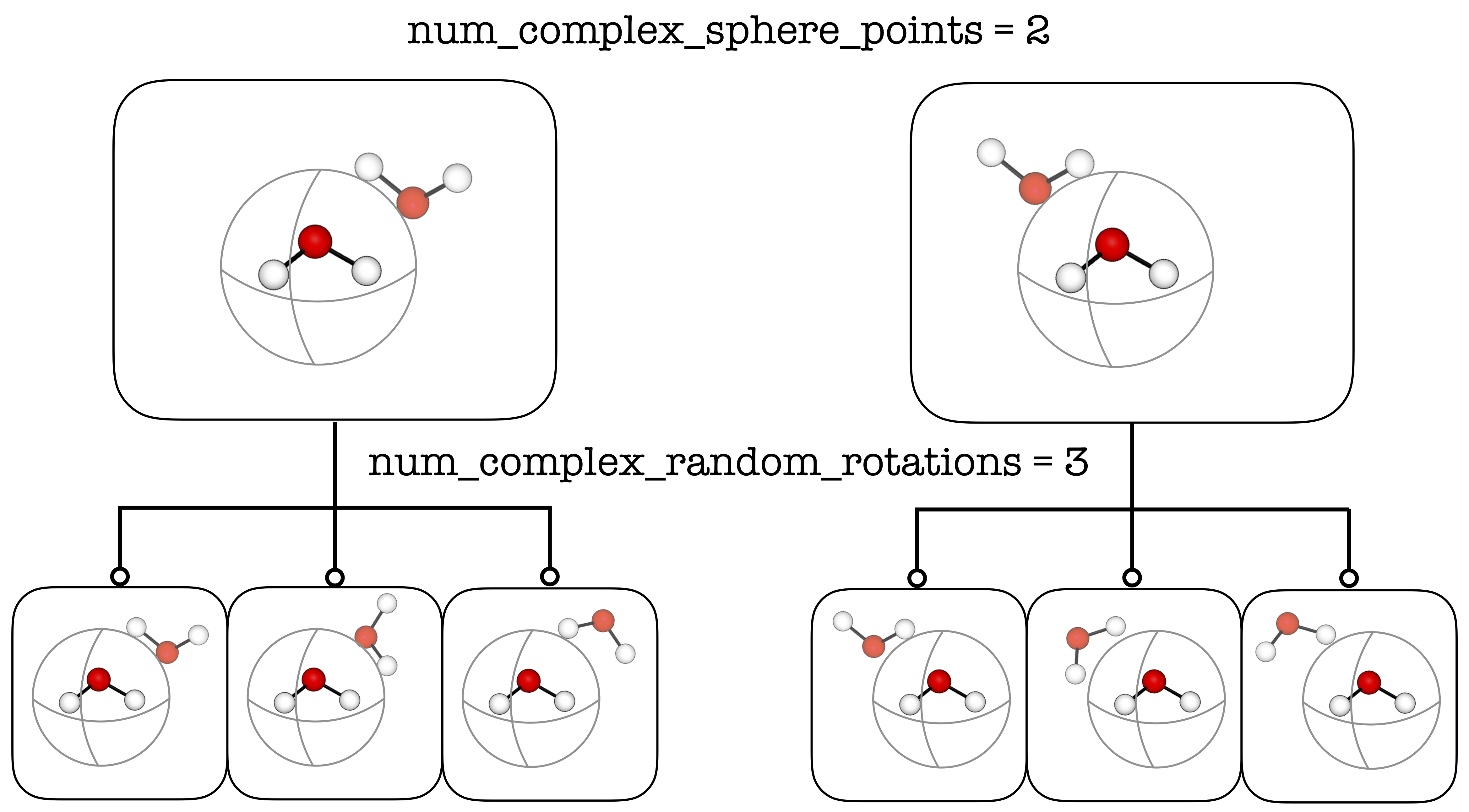
The parameters (num_complex_sphere_points and num_complex_random_rotations)
define the number of generated conformers, up to ade.Config.max_num_complex_conformers.
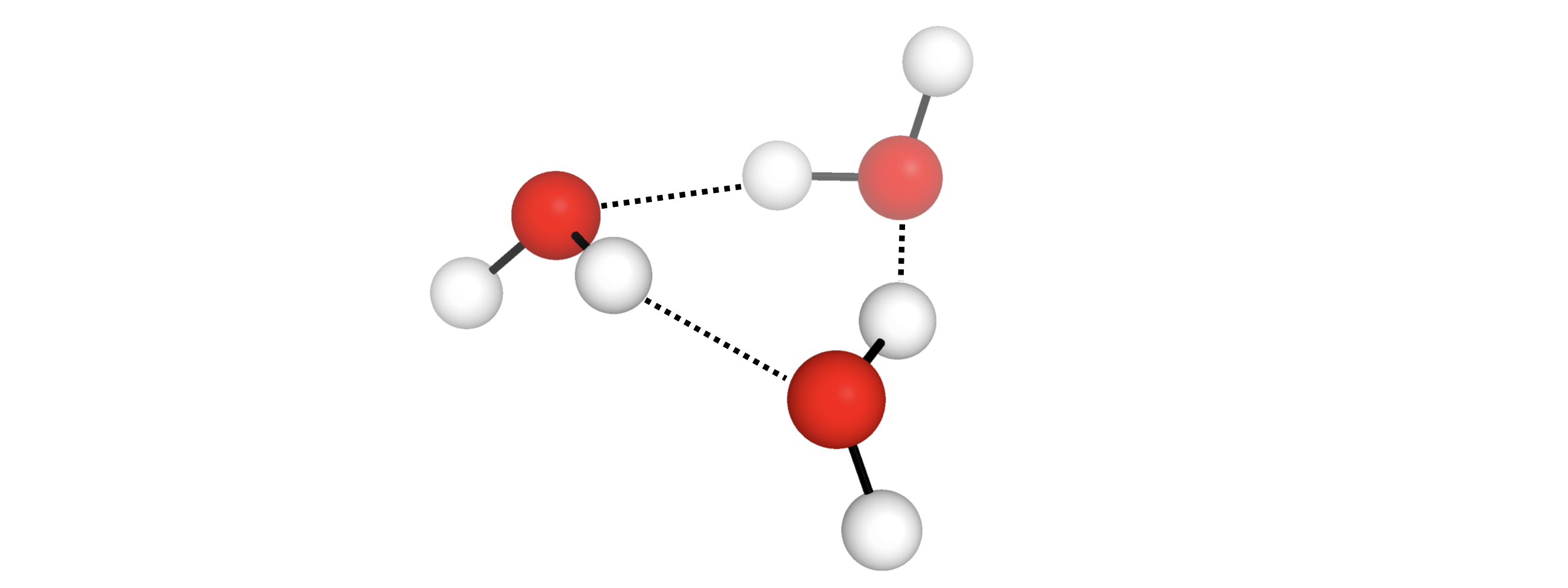
By default, autodE will exclude any conformers with differing connectivity, thus
may not generate any conformers of e.g. a [M(H2O)m]n+ system.
Complexes of systems with dative bonds can be generated by including
allow_connectivity_changes=True. For example, with a
[Na(H2O)3]+system:
import autode as ade
ade.Config.n_cores = 8
h2o = ade.Molecule(smiles="O")
na_ion = ade.Molecule(smiles="[Na+]")
# Initialise the [Na(H2O)3]+ complex and search 'conformers'
na_h2o_3 = ade.NCIComplex(na_ion, h2o, h2o, h2o)
na_h2o_3.find_lowest_energy_conformer(allow_connectivity_changes=True)
# Print .xyz files of all the generated conformers
for idx, conformer in enumerate(na_h2o_3.conformers):
conformer.print_xyz_file(filename=f"conf{idx}.xyz")
Out (visualised)