Conformer Generation#
autodE generates conformers using two methods: (1) ETKDGv2 implemented in RDKit and (2) a randomize & relax (RR) algorithm.
Butane#
To generate conformers of butane initialised from a SMILES string defaults to using ETKDGv2. The molecule’s conformers are a list of Conformer objects, a subclass of Species.
>>> import autode as ade
>>> butane = ade.Molecule(name='butane', smiles='CCCC')
>>> butane.populate_conformers(n_confs=10)
>>> len(butane.conformers)
2
where although 10 conformers requested only two are generated. This because by default there is an RMSD threshold used to remove identical conformers. To adjust this threshold
>>> ade.Config.rmsd_threshold = 0.01
>>> butane.populate_conformers(n_confs=10)
>>> len(butane.conformers)
8
For organic molecules ETKDGv3 is highly recommended, while for metal complexes the RR algorithm is used by default. To use RR for butane
>>> butane.rdkit_conf_gen_is_fine = False
>>> butane.populate_conformers(n_confs=10)
>>> for conformer in butane.conformers:
... conformer.print_xyz_file()
Out (visualised)
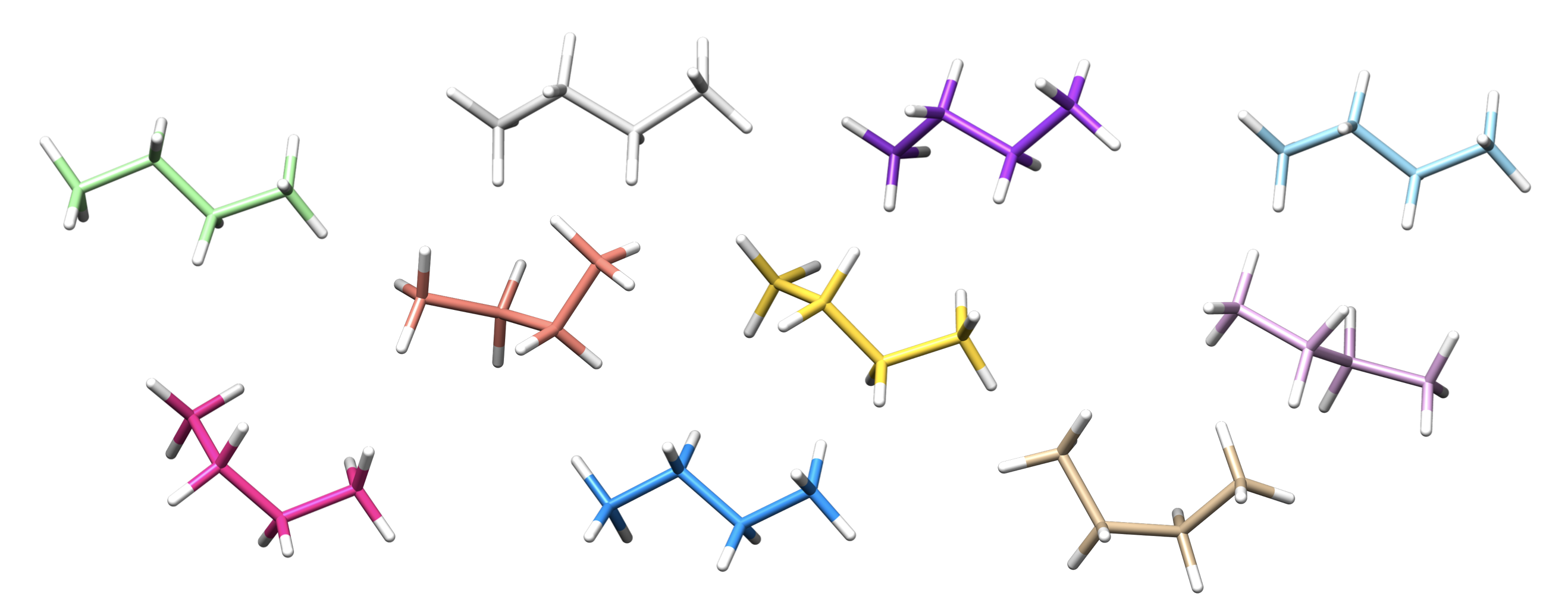
Note
RMSD used by the RR algorithm applies to all atoms and does not account for symmetry (e.g. methyl rotation)
Metal Complex#
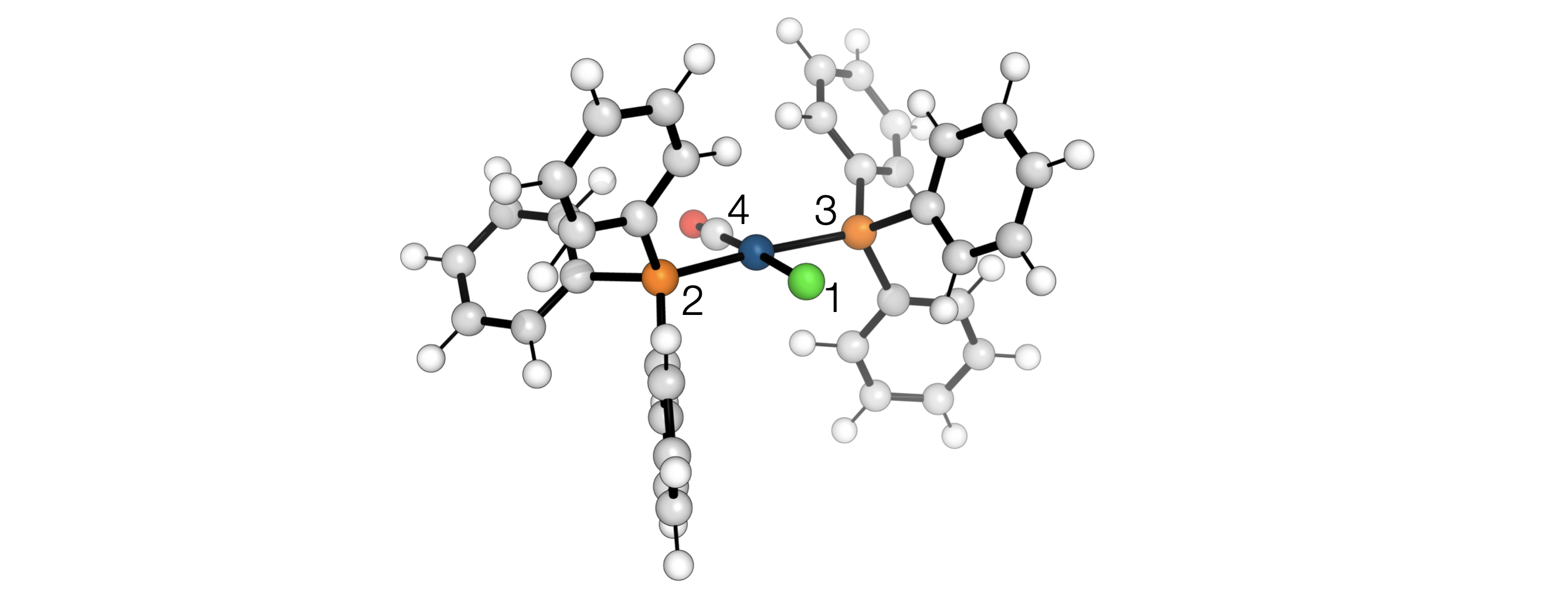
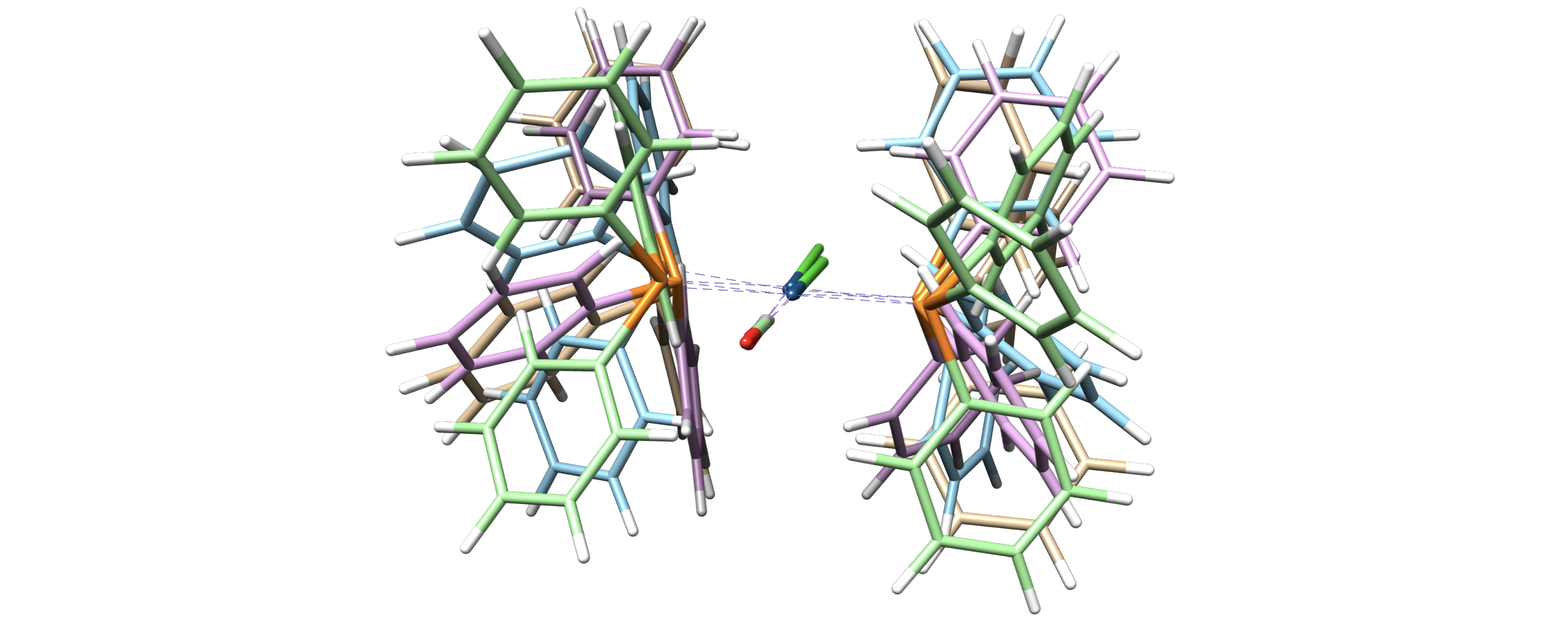
Arbitrary distance constraints can be added in a RR conformer generation. For example, to generate conformers of Vaska’s complex while retaining the square planar geometry
import autode as ade
from autode.conformers import conf_gen, Conformer
# Initialise the complex from a .xyz file containing a square planar structure
vaskas = ade.Molecule("vaskas.xyz")
# Set up some distance constraints where the keys are the atom indexes and
# the value the distance in Å. Fixing the Cl-P, Cl-P and Cl-C(=O) distances
# enforces a square planar geometry
distance_constraints = {
(1, 2): vaskas.distance(1, 2),
(1, 3): vaskas.distance(1, 3),
(1, 4): vaskas.distance(1, 4),
}
# Generate 5 conformers
for n in range(5):
# Apply random displacements to each atom and minimise under a bonded +
# repulsive forcefield including the distance constraints
atoms = conf_gen.get_simanl_atoms(
species=vaskas, dist_consts=distance_constraints, conf_n=n
)
# Generate a conformer from these atoms then optimise with XTB
conformer = Conformer(name=f"vaskas_conf{n}", atoms=atoms)
conformer.optimise(method=ade.methods.XTB())
conformer.print_xyz_file()
Out (visualised)