Species#
autodE provides Molecule classes built from a base Species class. A Species needs to be initialised from a name, set of atoms (or possibly None), charge and spin multiplicity
>>> import autode as ade
>>> species = ade.Species(name='species', atoms=None, charge=0, mult=1)
>>> species.n_atoms
0
Atoms are a list of Atom objects and can be used to initialise a species i.e.
>>> h2 = ade.Species(name='H2', charge=0, mult=1, atoms=[ade.Atom('H'), ade.Atom('H')])
>>> h2
Species(H2, n_atoms=2, charge=0, mult=1)
Atoms contain a coordinate as a numpy array (shape = (3,), initialised at the origin) and a few properties
>>> atom1, atom2 = h2.atoms
>>> atom1
Atom(H, 0.0000, 0.0000, 0.0000)
>>> atom1.coord
Coordinate([0. 0. 0.] Å)
>>> atom1.atomic_number
1
>>> atom1.atomic_symbol
'H'
>>> atom1.group
1
>>> atom1.period
1
Rotation and Translation#
Atoms can be translated and rotated e.g. to shift the first hydrogen atom from the origin along 1 Å in the x axis then rotate in the z-axis
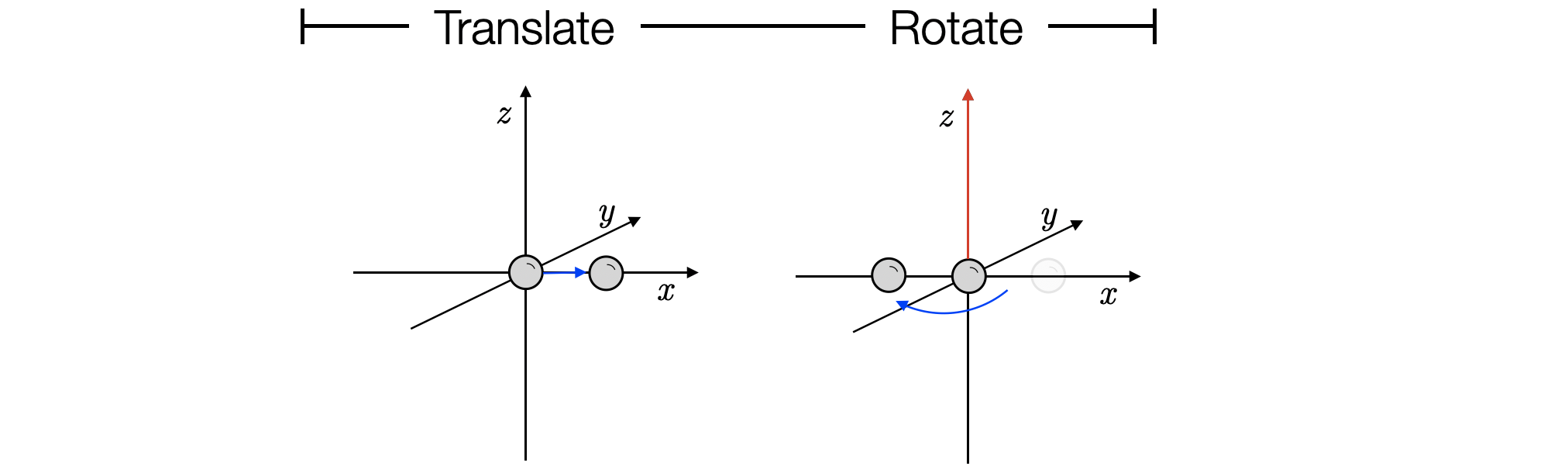
>>> vector = [1.0, 0.0, 0.0]
>>> atom1.translate(vector)
>>> atom1.coord
Coordinate([1. 0. 0.] Å)
To rotate this atom 180° (π radians) in the z-axis at the origin
>>> atom1.rotate(theta=3.14159, axis=[0.0, 0.0, 1.0])
>>> atom1.coord
Coordinate([-1. 0. 0.] Å)
Note
Rotations are performed anticlockwise
Translations and rotations are performed in place so the h2 atoms are modified
>>> h2.atoms
Atoms(n_atoms=2, [Atom(H, -1.00, 0.00, 0.00), Atom(H, 0.00, 0.00, 0.00)])
Distances#
Distances between atom pairs can be calculated, where atoms are indexed from 0. To calculate the bond length for this species
>>> h2.distance(0, 1)
Distance(1.0 Å)
Distances support conversion into other units (bohr, nano/pico-meters), as well as all standard mathematical operations
>>> h2.distance(0, 1).to('a0')
Distance(1.88973 bohr)
>>> 2 * h2.distance(0, 1)
Distance(2.0 Å)
Angles#
Bond angles can be calculated between three atoms. For example, in a water molecule
>>> h2o = ade.Species(name='H2O', charge=0, mult=1,
... atoms=[ade.Atom('H', x=-1.0),
... ade.Atom('O'),
... ade.Atom('H', x=0.25, y=0.97)])
>>> h2o.angle(0, 1, 2).to('degrees')
Angle(104.45247 °)
Similarly, dihedral angles are available using Species.dihedral.
Solvents#
Species also support a solvent, which need not be specified for a species in the gas phase
>>> h2.solvent is None
True
For example, to initialise a fluoride ion in dichloromethane
>>> f = ade.Species(name='F-', charge=-1, mult=1,
... atoms=[ade.Atom('F')],
... solvent_name='DCM')
>>> f.solvent
Solvent(dichloromethane)
Given a solvent name string a Solvent is added as an attribute to the species. A Solvent contains a set of aliases and names of the implicit solvent in different electronic structure theory packages e.g.
>>> f.solvent.g09
'Dichloromethane'
>>> f.solvent.xtb
'CH2Cl2'
Species from Files#
Species may be initialised from xyz files using the io module
>>> from autode.input_output import xyz_file_to_atoms
>>> methane = ade.Species(name='CH4', charge=0, mult=1,
... atoms=xyz_file_to_atoms('methane.xyz'))
>>> methane
Species(CH4, n_atoms=5, charge=0, mult=1)
Note
Only .xyz files are supported currently. Other molecular file formats can be converted to .xyz with openbabel.