Changelog#
1.4.5#
Functionality improvements#
Adds a boolean option for calculating single points refinements
Bug Fixes#
Fixes coordinate extraction in some G16 output files
Fixes loading old mlptrain .npz files
Fixes compatability with RDKit 2025.03.2
Usability improvements/Changes#
Drops Python 3.8 support
Catches conformer calculation exceptions
1.4.4#
Functionality improvements#
Improved constrained optimisation (
CRFOptimiser) and handling of multiple constraintsAdds compatability with numpy v2.0
Improved implementation of the RFO-TRM (
QAOptimiser) optimiser that can handle constraintsAdded static internal back-transform and damping for faster and easier DIC to Cartesian coordinate transformation
Bug Fixes#
DIC to Cartesian transform will now always use
PIC.close_to()to ensure steps along dihedral have the smallest change, even after back-transform is complete
Usability improvements/Changes#
Optimiser convergence criteria have been improved to consider energy change, RMS and max. gradient and step sizes.
1.4.3#
Functionality improvements#
DHS and DHS-GS can now switch between two step sizes
Peak detection in bracket methods now uses cubic polynomial fit with energies and gradients instead of only projecting gradients
Bug Fixes#
Fixes no solvent being added in QRC calculations
Fixes cases where .xyz files are printed with no space between coordinates when the coordinate value is large.
Fixes DHS and DHS-GS methods ignoring number of cores
Usability improvements/Changes#
Added consistent aliases for double dagger across all energies in
autode.reaction.deltaOptimiser trajectories are saved on disk instead of keeping completely in memory
Hessian updates the refactored into
OptCoordinatesclass
1.4.2#
Functionality improvements#
Replaces hard-coded derivatives for primitive internal coordinates with automatic differentiation
More comprehensive primitive internal coordinate generation
Adds the capacity to handle linear molecules in internal coordinate system
Bug Fixes#
Fixes triangular rings being incorrectly treated as dihedral angles
Usability improvements/Changes#
Faster import of autode package by lazily loading matplotlib
ORCA output files copied after a calculation are configurable
1.4.1#
Functionality improvements#
Adds the thermochemistry method from A. Otlyotov, Y. Minenkov in https://doi.org/10.1002/jcc.27129
Adds the improved Elastic Image Pair (i-EIP) method for double-ended transition state search
Adds a
autode.Species.solvent_nameproperty and setter for setting solvents from a stringEnables reloading molecules from xyz files with their previously defined charge/multiplicity/solvent
Bug Fixes#
Fixes Hessian extraction in some G16 output files
Fixes large step sizes in DHSGS
Fixes the ability to define both a SMILES string and a xyz file without raising an exception
1.4.0#
Functionality improvements#
Adds
temporary_config()context manager for temporary configuration changesAdds a
ForceConstantvalueAdds a more robust hybrid RFO-TRM geometry optimiser with trust radius update and a feature to detect oscillation and damp it
Adds Dewar-Healy-Stewart (DHS) method and its variant DHS-GS to find transition states from initial and final geometries
Usability improvements/Changes#
Adds full usability of autodE on Windows, including parallelisation with
lokyOptional timeout for graph isomorphism test in Windows, turned on by
Config.use_experimental_timeout=True(default behaviour kept for Linux/macOS)The electronic temperature and the version of parameterisation for xTB calculations are made configurable
A NEB
Imagenow derives from aSpeciessuperclassModifies NEB
Imageconstructor to be formed from an imageDefines named constructors (
from_end_points(...),from_list(...)) forNEBRemoves
NEB().contains_peak()in favour ofNEB().images.contains_peakModifies the
CImagesconstructor to ensure it’s constructed from anImagesinstanceRemoves
NEB.get_species_saddle_point()in favour ofNEB.peak_speciesDefines named constructors (
from_end_points(...),from_list(...)) forNEBRemoves
mol_graphs.get_atom_ids_sorted_type()
Bug Fixes#
Fixes pickling issue with
autode.config.Configon Windows and in multiprocessing spawn for Linux/macOSFixes RFO Hessian update bug
Fixes QChem not using
Config.max_core
1.3.5#
Usability improvements/Changes#
autode.value.ValueArray.to()now defaults to copying the object rather than inplace modification
Functionality improvements#
Adds a
to_method toautode.value.ValueArrayfor explicit inplace modification of the array
Bug Fixes#
Fixes
ERRORlogging level being ignored from environment variableAUTODE_LOG_LEVELFixes
autode.values.Valueinstances generating items with units on division, and throw a warning if multiplyingFixes the printing of cartesian constraints in XTB input files, meaning they are no longer ignored
Fixes
Hessianinstances changing units when normal modes are calculatedFixes an incorrect alias for
ev_per_ang
1.3.4#
Feature additions.
Usability improvements/Changes#
Throw useful exception for invalid
ade.Config.ts_template_folder_path
Functionality improvements#
Adds
ade.transition_states.TransitionState.from_speciesmethod to construct transition states from a species or moleculeAdds
autode.Reaction.save()andautode.Reaction.load()to save and reload a reaction stateAdds saving checkpoints of a reaction during
autode.Reaction.calculate_reaction_profile
Bug Fixes#
Fixes calculation
clean_up()failing with a null filename
1.3.3#
Bugfix release.
Functionality improvements#
Adds skipping Hessian re-evaluation when using autodE optimisers if a molecules has a Hessian calculated at the same level of theory
Adds a Hessian recalculation frequency to
autode.optimisers.PRFOptimiserImproves the default step size for TS optimising to be consistent with the ORCA default
Bug Fixes#
Adds checking of SMILES-defined charge against the user-specified value
Fixes
autode.optimisers.CRFOptimiserbuilding incomplete internal coordinates for partially or completely fragmented molecules
1.3.2#
Bugfix release.
Usability improvements/Changes#
Removes
autode.geom.get_distance_constraintsin favour of a better named methodTSBase().active_bond_constraints
Bug Fixes#
autode.transition_states.ts_guess.TSguess.from_speciesnow inherits solvent from the speciesFixes a possible race condition in I/O for XTB conformer optimisations
1.3.1#
Bugfix release.
Bug Fixes#
Fixes behaviour of
autode.utils.work_in_tmp_dirandautode.utils.work_indecoratorsFixes an exception being raised when
autode.Calculation.clean_upis called with a method that doesn’t implement external I/OFixes autodE driven optimisations skipping execution when the input but not name changes
1.3.0#
Optimisation features, graph assignment improvements and bugfixes.
Usability improvements/Changes#
Defines dummy atoms to have zero covalent and vdW radii
Renames
Method().availabletoMethod().is_availableRemoves
autode.bonds.get_ideal_bond_length_matrixandautode.bonds.get_avg_bond_lengthRemoves
autode.geom.rotate_columnsModifies the names of most optimiser classes e.g.
autode.opt.optimisers.PRFOOptimiser->PRFOptimiserSimplifies initialising a
autode.calculations.Calculationby:Requiring constraints to be attributed to a molecule
Removing the
bond_ids_to_addargument and using the labeled graph instead (active edges)Removing the
other_input_blockargument and appending to the keywords instead
Removes
autode.calculations.Calculation.print_final_output_linesin favour of a method oncalculation.outputMakes many methods in
autode.calculations.CalculationprivateDeprecates all
autode.calculations.Calculation.get_<method>methods in favour of setting properties of the input moleculeReturns
Nonerather than rasies exceptions when calling the (deprecated) calculation get methods, to be consistent with …get_energyAdds an
autode.wrappers.keywordspackage to improve file structureRemoves any exceptions on calling
.run()on an optimiser instance where the system has no degrees of freedomRemoves support for Python < v3.8
Tweaks the default ORCA TS optimisation keywords to be more conservative, i.e. slower and more accurate
Functionality improvements#
Adds a
autode.atoms.Atom.covalent_radiuspropertyAdds a
autode.atoms.Atoms.eqm_bond_distancemethod for the equilibrium bonded distance between two atomsAdds vibrational frequency scaling through both
autode.Config.freq_scale_factorand a default value in wrapped functional keywordsAdds a much more robust constrained rational function constrained optimiser in delocalised internal coordinates (DIC)
Adds bond angle and dihedral primitive coordinates which can form part of the DIC set
Improves the back transformation
Adds an optional callback argument to
autode.opt.optimisers.base.Optimiserfor running custom functions after every optimisation stepAdds the ability to save/reload an
autode.opt.optimisers.NDOptimiserinstance to/from a fileAdds a solvent attribute to a
autode.transition_states.transition_state.TransitionStateconstructorAdds functionality to partition a nudged elastic band into images where the maximum atom-atom distance between images is below a threshold
Adds a sequential adapt+NEB TS finding method where a pure adapt. path fails to generate a geometry close enough to the TS for a successful TS optimisation
Bug Fixes#
Fixes variable harmonic frequencies (<2 cm-1 differences) due to projection vectors becoming close to rotational axes
Fixes the extraction of atomic partial charges from ORCA output files
Fixes gradients and Hessians not being reset on a molecule where the coordinates change
Fixes unhelpful exception when calculating thermochemistry with EST methods without implemented “get_hessian” methods
See the table below for a quick benchmark of constrained optimisations in autodE compared to ORCA. In all cases the structures were generated from SMILES strings (RDKit) and optimised with a single constraint on the (0,1) distance of +0.1 Å from its current value.
Molecule |
autodE |
ORCA |
|---|---|---|
C7H12 |
6 |
7 |
C3H7 |
8 |
17 |
C4H6 |
3 |
5 |
CClH3 |
3 |
4 |
C2H3O2 |
3 |
7 |
C2FH5 |
3 |
5 |
C4H6O2S |
6 |
11 |
1.2.3#
Minor functionality improvements and bugfixes.
Usability improvements/Changes#
All exceptions now inherit from a base
autode.exceptions.AutodeExceptionFixes a typo in
autode.exceptions.UnsupoportedCalculationInputAdds documentation explaining the intention of each exception in
autode.exceptionsMolecular graphs are now ‘laziliy-loaded’ i.e. generated once when the property is accessed
Functionality improvements#
Adds the ability to define atom classes for molecules in turn allowing for identity reactions to be calculated
Bug Fixes#
Fixes clashing names for a reaction initialised explicitly from molecules without defined names
1.2.2#
Bugfix release.
Bug Fixes#
Fixes output redirection from XTB calculations resulting in missed lines on Mac
1.2.1#
Bugfix release.
Functionality improvements#
Adds
autode.mol_graphs.MolecularGraph(subclass ofnetworkx.Graph) with aexpected_planar_geometrymethodAdds an
are_planarmethod toautode.atoms.Atoms
Bug Fixes#
Fixes a bug in assigning reasonable geometries which, in turn, could lead to skipped single point energy evaluations
1.2.0#
Adds optimisation algorithms experimental explicit solvation, improves potential energy surface module as well as an array of usability improvements.
Usability improvements/Changes#
Adds more type hints and documentation
Updates the TS template saved in the default template library
Adds a setter for
autode.species.Species.solventsomol.solvent = 'water'will assign aautode.solvent.SolventRemoves
autode.calculation.CalculationInput.solventas an attribute in favour of using the molecule’s solventRemoves
autode.calculation.get_solvent_namein favour of a molecule checkRemoves
autode.species.molecule.reactant_to_productin favour of ato_product()method forautode.species.molecule.Reactant(and likewise with a Reactant)Removes partially implemented
autode.species.molecule.SolvatedMoleculeandautode.species.complex.SolvatedReactantComplexas the type of solvation (implicit/explicit) should be a property of the solvent and not the moleculeRemoves
autode.reactions.Reaction.calc_deltaXXXin favour ofautode.reactions.Reaction.delta()Refactors classes to place constructors at the top
Removes
autode.values.PlottedEnergyas an estimated attribute is useful for all energies, not just those that are plottedRemoves
autode.reactions.Reaction.find_lowest_energy_tsas the function is not well named and can be replaced by aautode.reactions.Reaction.tspropertyAdds
autode.transition_states.TransitionStatesas a wrapper for TSs, much likeautode.conformers.ConformersUpdates
autode.solvent.solvents.get_solventto require specifying either an implicit or explicit solventImproves validation of distance constraints and adds invariance to the key order i.e.
autode.constraints.distance[(0, 1)] == autode.constraints.distance[(1, 0)]Removes
autode.KcalMolandKjMoland enables a reaction to be plotted using a string representation of the units.Allows for keywords to be set using just a list or a string, rather than requiring a specific type
Changes
autode.wrappers.keywords.Keyword.has_only_nameto a propertyModifies the constructor of
autode.species.molecule.Moleculeto allow for a name to be specified when initialising from a .xyz fileModifies
autode.calculation.Calculation.get_energyto raise an exception if the energy cannot be extractedAdds a runtime error if e.g.
autode.calculation.Calculation.get_energyis called on a calculation that has not been runSkips low-level adaptive path searching if the high and low-level methods are identical (when XTB or MOPAC are not installed)
Adds a default set of low-level single point keywords
Adds a flag to override exiting a reaction profile calculation when association complexes are generated
Adds a check that a calculation isn’t going to exceed the maximum amount of physical memory on the computer
Functionality improvements#
Adds a selection of molecule optimisers to locate minima and transition states
Refactors
autode.smiles.anglesto use unique class names (preventing overlap with e.g.autode.values.Angle)Adds a
autode.solvent.Solvent.dielectricproperty for a solvent’s dielectric constantAdds a
autode.solvent.Solvent.is_implicitpropertyAdds methods (e.g. translate and rotate) to
autode.point_charges.PointChargeAdds checking that both high and low-level electronic structure methods are available before running
autode.reaction.Reaction.calculate_reaction_profileorcalculate_reaction_profileAdds a more robust explicit solvation generation (
autode.species.molecule.Molecule.explicitly_solvate())Removes criteria on using a TS template with large distance differences between the structure and the template in favour of running sequential constrained optimisations to the required point
Rewrites
autode.pesinto a consistent module while maintaining much of the functionality. Simplifies the interfaceAdds a QChem electronic structure method wrapper
Adds
autode.species.Species.calc_hessianto calculate either an analytic or numerical Hessian (in parallel)Adds image dependent pair potential (IDPP) relaxation improved interpolated geometries
Adds
autode.hessians.HybridHessianCalculatorto calculate numerical Hessians at two levels of theory
Bug Fixes#
Updates the TS template saved in the default template library
Reloads output file lines from a failed then re-run calculation
Fixes Hessian extractions from some Gaussian output files
1.1.3#
Usability improvements
Usability improvements/Changes#
Improves consistency and behaviour of
calc_thermomethod of a species, allowing for keywords and non-run calculationsAllows for a non-fork multiprocessing ‘start_method’
1.1.2#
Bugfixes
Usability improvements/Changes#
Fixes typo in
autode.exceptions.ReactionFormationFalied
Bug Fixes#
Fixes a bug where rings containing mostly double bonds failed to build with
autode.smiles.builder.BuilderFixes using XTB as a high-level method with the xtb-gaussian wrapper (thanks @kjelljorner)
1.1.1#
Documentation and typing hints
Usability improvements/Changes#
Adds typing to user-facing functions
Adds
autode.config.locationto easily locate the core configuration file for permanent editingUpdates documentation for readability
Ensures units are kept if constructing a
Valuefrom aValue(i.e.Value(x), whenxis aValue)
Functionality improvements#
Changes
Keywordto an abstract base classImproves speed of
Speciesrotation (numpy rather than a Python for loop)
Bug Fixes#
Fixes bug where NCI conformers were generated with the same name thus did not optimise uniquely (introduced in v.1.1.0)
1.1.0#
API improvements that broadly maintain backwards compatibility.
Usability improvements/Changes#
Adds more argument and return types
Changes
AtomCollection.atomsto a property for more flexible sub-classingChanges
ElectronicStructureMethod.doi_strandKeyword.doi_strto propertiesAdds interpretable
repr(Species)Species.energiesis zeroed when theSpecies.atomsare reset or changeSpecies.energyis a property of the last computed energy on that speciesSpecies.is_linearnow uses an angle tolerance to determine linearity, which is slightly tighter than the previous float-based toleranceRemoves
CalculationOutput.set_linesin favour of a cached file_lines property to avoidset_file_lines()Removes
CalculationOutput.get_free_energy()in favour ofSpecies.free_energyonce a Hessian is set for a molecule and similarly withCalculationOutput.get_enthalpy()Removes
CalculationOutput.get_imaginary_freqs()(nowSpecies.imaginary_frequencies) andCalculationOutput.get_normal_mode_displacements()(nowSpecies.normal_mode())Species.imaginary_frequenciesnow returnsNonerather than an empty list for a species without any imaginary frequencies, to be consistent with other propertiesChanges
CalculationOutput.terminated_normally()to a property (CalculationOutput.terminated_normally)Removes
Reaction.find_complexesin favour of setting the reactant and product complexes dynamically, unlessReaction.calculate_complexesis called to find association complexesTweaks the default relative tolerance on bonds to account for M-X agostic interactions lengthening bonds
Enables
Species.atomsto be added, even if they are NoneImproved atom setting of
Complex.atomsChanges
Complex.get_atom_indexes()toComplex.atom_indexes()Changes
Complex.moleculesto a private attribute as the atoms/energy/gradient is not propagatedAllows for
Species.translate()andSpecies.rotate()to be called using vectors as lists or tuples rather than just numpy arraysModifies
get_truncated_complex()toget_truncated_species()and changes the return type to a species to reflect a possibly different molecular composition of the complexImproves peak checking in adaptive path TS guess generation
Removes
autode.atoms.get_thing()functions, in favour ofAtom.thingRaises an exception if a single point energy evaluation fails to execute successfully
Removes
autode.conformers.conformer.get_conformer()in favour of a more flexibleautode.conformer.ConformerconstructorAdds
Species.constraintsthat are used in optimisations (still available inCalculationinitialisation)Adds
Conformersto enable parallel electronic structure calculations across a set of conformersImproves readability of pruning of conformers based on RMSD and energy thresholds
Functionality improvements#
Adds angle and dihedral angle properties to an
AtomCollectionImproves and adds more
UnitdefinitionsAdds
ValueandValueArraybase classes for energies, gradients etc. These allow for implicit (1 Hartree == 617.509 kcal mol-1) comparisons and explicit conversion (1 Hartree).to(‘kcal’)Adds further conversion factors to
ConstantsAdds
Species.energiesas a container of all energies that have been calculated at a geometryAdds
Keywords.bstringas a ‘brief’ summary of the keywords e.g. PBE0/def2-SVP and are associated with anEnergy(a type ofValue)Improves quick reaction coordinate characterisation of TSs by providing a maximum atomic displacement for improved initial structures
Adds Hessian diagonalisation to obtain normal modes with and without translation and rotation projections for linear and non-linear molecules
Adds
Species.weightandSpecies.massas equivalent properties for the molecular weightImproves dihedral sampling in molecule generation
Adds
atoms.remove_dummy()to remove all dummy atoms from a setEnables different force constants to be used in XTB constrained optimisations (
Config.XTB.force_constant, which setswrappers.XTB.XTB.force_constant)Adds
Solvent.copy()Adds
Species.reorder_atoms()to reorder the atoms in a species using a mappingAdds
Config.ORCA.other_input_blockto allow for a block of input to be printed in all ORCA input filesChanges the loose optimisations to only use a maximum of 10 iterations. This is based on an analysis of 3500 ORCA
optimisations, which plateaus quickly:
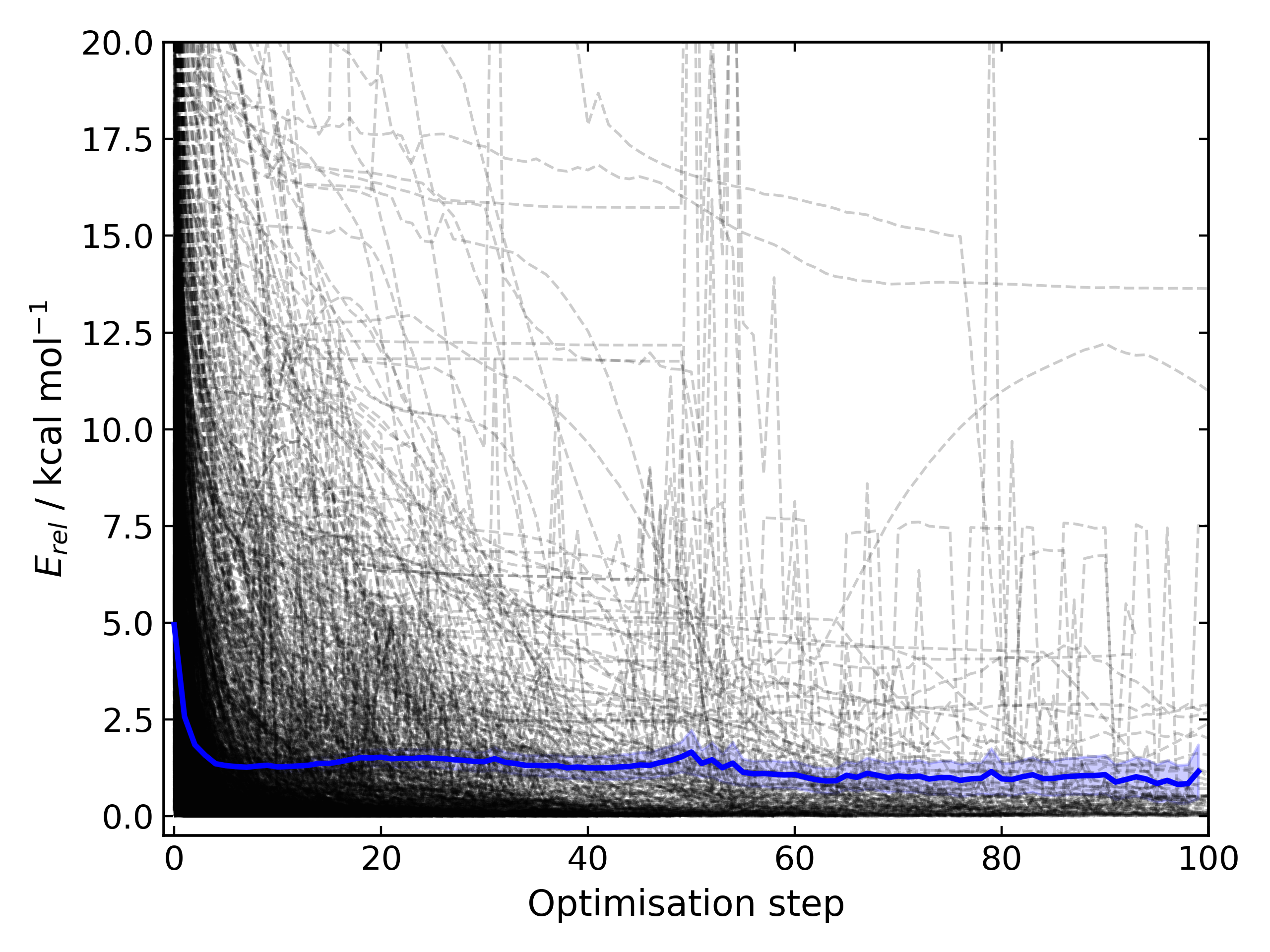
suggesting a value of 10 is a appropriate. This will be system dependent and need increasing for large/flexible systems. For path optimisations loose optimisations use a maximum of 50 cycles.
Bug Fixes#
Skips conformers with no atoms in finding unique conformers
Corrects benchmark TS location for the Grubbs metathesis example, where the reactant complex is bound
Fixes possible zero distance constraint for a single atom
Fixes spin state definition for XTB calculations
Fixes possible override of a constructor-defined spin state by the SMILES parser
1.0.5#
Bugfix release
Bug Fixes#
Saves transition state templates with correct atom labels
1.0.4#
Bug fixes in SMILES parser and 3D geometry builder from 1.0.3.
Usability improvements#
Improves doc strings
Throws interpretable error when calling
find_tsswithoutreaction.reactantset
Functionality improvements#
SMILES strings with >9 ring closures are parsed correctly
cis-double bonds in rings no longer minimise with constraints, which is a little faster
Bug Fixes#
Tweaks repulsion parameters in minimisation to build fused rings
Enables SMILES parsing with “X(…)1” branching
Fixes spin multiplicity for odd numbers of hydrogens
Improves ring closure 3D build
Fixes incorrect implicit valency for aromatic heteroatoms
Improves metal finding in SMILES strings with regex
Corrects atom type for sp2 group 16 elements
Fixes dihedral rotation with atoms not close to any other
1.0.3#
A minor API revision from 1.0.2 but adds C++ extension which should be extensible to further developments of fast C-based code.
Usability improvements#
autode.Species()inherit from aAtomCollection()base class for more flexibilityautode.Constantsattributes have more readable names (while retaining backwards compatability)autode.geom.length()as an explicit alias ofnp.linalg.normhas been removedautode.input_output.xyz_file_to_atoms()throws more informative errorsautode.mol_graphs.make_graph()throws NoAtomsInMolecule for a species with no atomsspecies.formulaandspecies.is_explicitly_solvatedare now a propertiesautode.smiles.parserhas been rewritten & is (hopefully) a more robust SMILES parser
Functionality improvements#
Metal complex initial geometries can now be generated with the correct stereochemistry
Macrocycles default to an autodE builder that conserves SMILES stereochemistry (RDKit#1852)
species.coordinatescan be set from either 3xN matrices or 3N length vectorsautode.Atom()`s have :code:.group`.periodand.tm_rowproperties referring to their location in the periodic tableautode.atoms.PeriodicTableaddedspecies.bond_matrixadded as a property and returns a boolean array for interactions between all atom pairs
Bug Fixes#
reaction.calculate_complexes()callsreaction.find_complexes()if needed thus can be called in isolation
1.0.2#
Usability improvements#
Effective core potentials can now be specified in
Keywords()ORCA fitting basis sets now default to def2/J, which should be smaller but as accurate as AutoAux
Molecule initialisation from a .xyz file now checks for an odd number of electrons. For example,
Molecule('H_atom.xyz')will raise aValueErrorbutMolecule('H_atom.xyz', charge=1)orMolecule('H_atom.xyz', mult=2)are acceptable
Functionality improvements#
atom.atomic_numberhas been added as an atom attributeatom.atomic_symbolis a more intuitive alias foratom.label
1.0.1#
Usability improvements#
Molecular complexes can now be initialised with a reasonable geometry
Complex(..., do_init_translation=True)
Functionality improvements#
species.radiushas been added as an approximate molecular radius (in Angstroms, excluding VdW radii)
Bug Fixes#
Final breaking bond distances are now the minimum of the product X-Y distance if present in the product, or 2x the distance. This is required for breaking bonds that cross a ring.
Neighbour lists for comparing possibly equivalent bond rearrangements are now compared using a sorted list
1.0.0#
The first stable release! Mostly documentation updates from v.1.0.0b3 with the package now being conda-install-able.
Usability improvements#
More documentation
Functionality improvements#
XTB wrapper now supports v. 6.4 (and hopefully higher)
Thanks to Joe, Alistair, Matina, Kjell, Gabe, Cher-Tian amongst others for their invaluable contributions.
1.0.0b3#
This version brings several major changes and in some instances breaks backwards compatibility, but does feature significant improvements in speed and accuracy for finding transition states.
Usability improvements#
species.get_distance(i, j)is nowspecies.distance(i, j)species.set_atoms(new_atoms)is now properly handled with a setter sospecies.atoms = new_atomswill set the new atomsspecies.n_atomsis more robustspecies.get_coordinates()is nowspecies.coordinates, returning a numpy array copy of the species coordinates (Nx3 in Å)species.centre()will translate a species so it’s coordinate centroid lies at the originPBE0/def2-SVP is now the default ‘low opt’ method (keywords.low_opt) with loose optimisation. Path exploration uses this method, thus it needs to be very close to the ‘opt’ level
Functionality improvements#
1D, 2D potential energy surface scans and nudged elastic band (NEB) methods to generate TS guesses from reactants have been replaced by an adaptive path search which seems to be very efficient for generating initial paths
For the prototypical SN2 between fluoride and methyl chloride the relaxed PES (PBE0-D3BJ/ma-def2-SVP/CPCM(water)) is
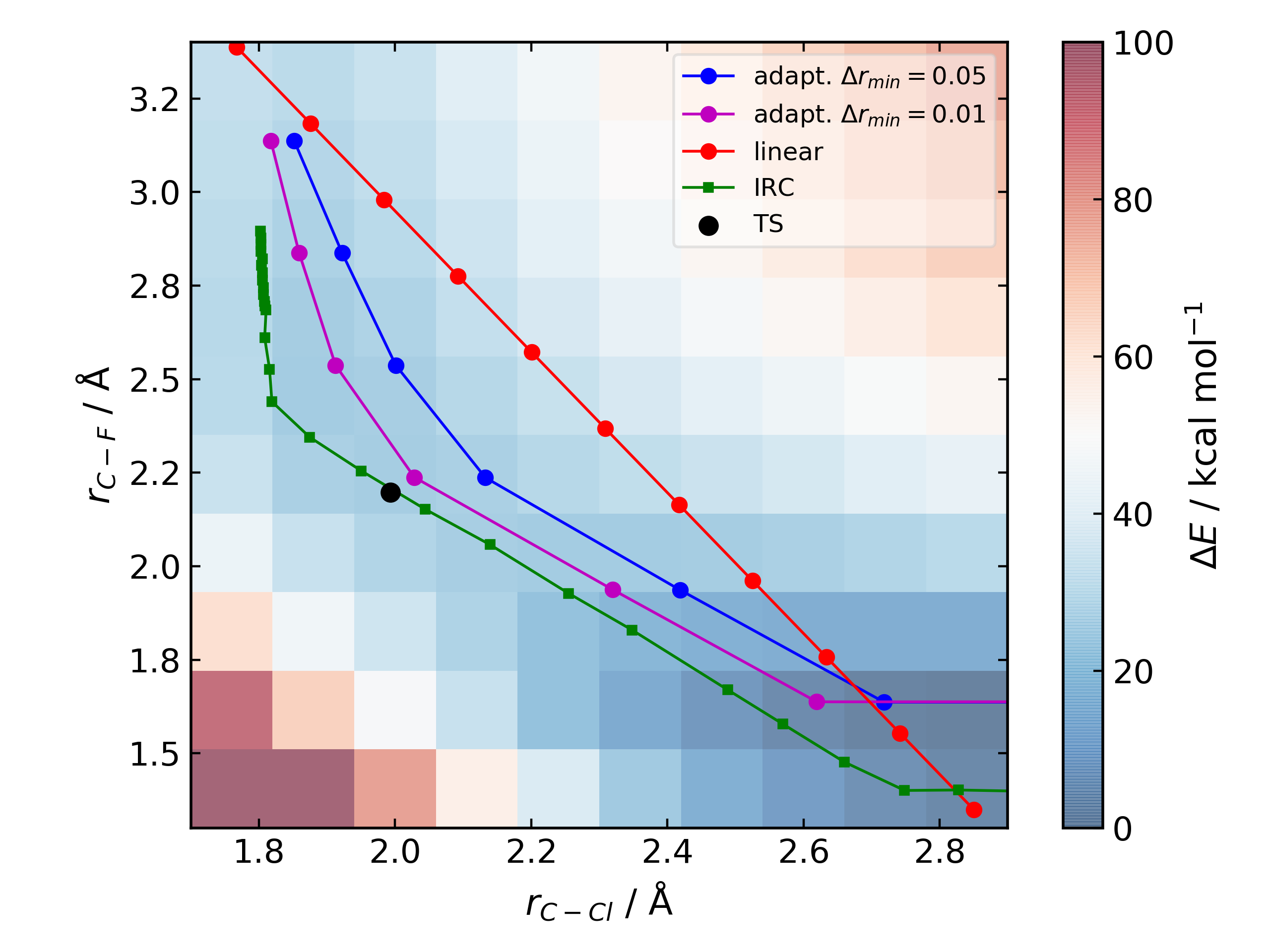
where the previously employed linear path (red) is compared to the adaptive scheme (blue, purple) and the ‘true’ intrinsic reaction coordinate. With a small minimum step size a path very close to the MEP is traversed with a very small number of required constrained optimisations. This enables NEB relaxations to be skipped and the associated limitations (corner cutting, oscillating path, optimisation in Cartesian coordinates) avoided. This exploration is essential when a linear path over multiple bonds leads to rearrangements, e.g. an (E2) elimination reaction the comparison for the linear, adaptive and IRC paths are shown below
(CI)-NEB with adaptive force constant has been added
Initial path exploration from reactants is performed at the ‘low_opt’ level with a final breaking bond distance below.
Previous implementations made use of a 1.5 Å additional shift for uncharged reactions and 2.5 Å for charged, this however lead to possible final C-H distances of ~3.6 Å and steps into unphysical regions. 1.0.0b3 uses an estimate based on the distance where the bond is mostly broken, as below
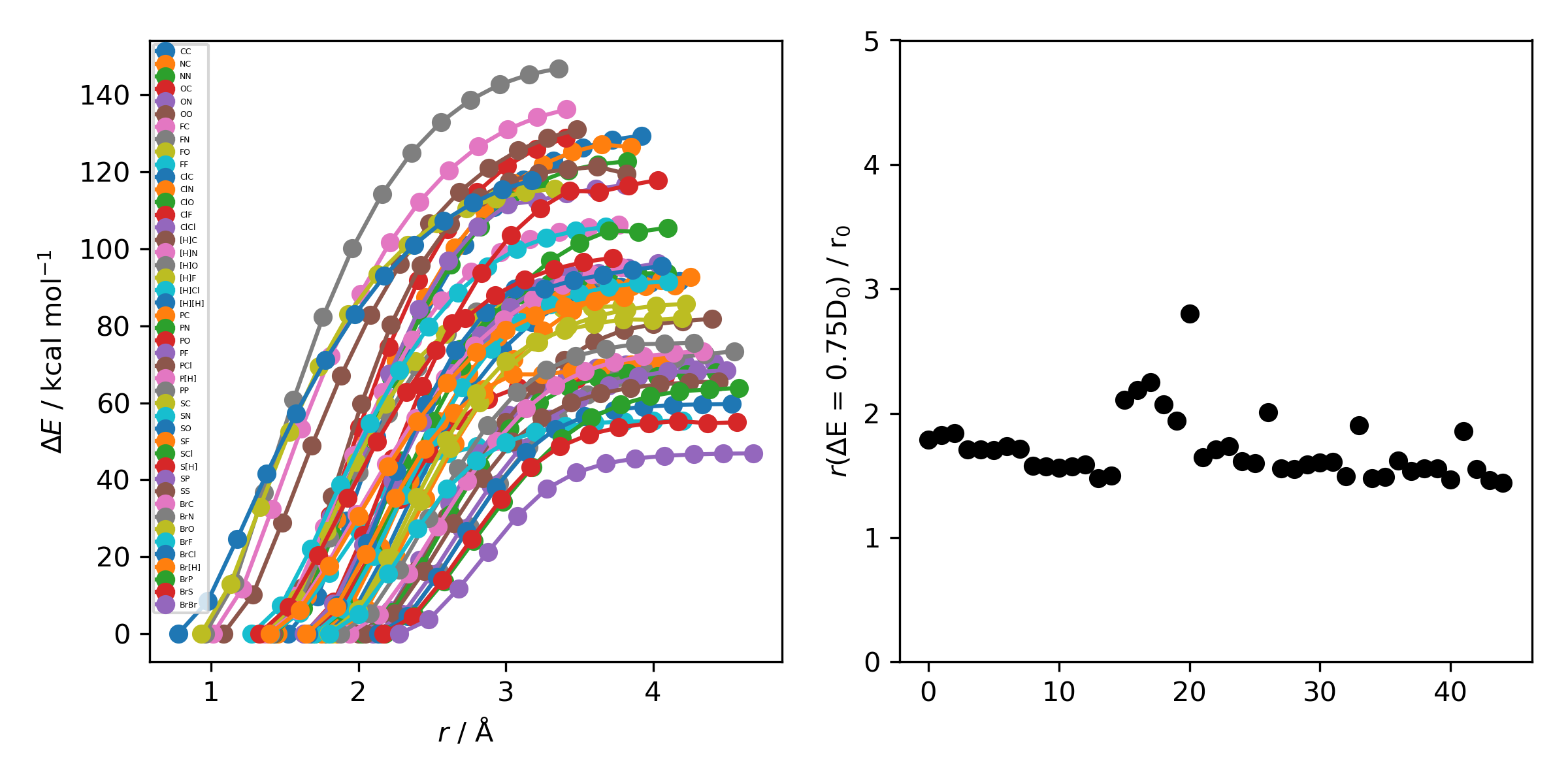
where X-Y corresponds to a molecule e.g. C-C with the appropriate hydrogens added then the BDE curve calculated at the GFN2-XTB level of theory. A multiplier of ~2 affords a ‘mostly broken bond’ (i.e. the distance at 3/4 of energy of the broken bond).
There is now a heuristic used to skip TSs that go via small rings (3, 4-membered) if there is a >4-membered equivalent (
ade.Config.skip_small_ring_tss)
Bug Fixes#
Calculations are now unique based on constraints, so NEB calculations executed in the same directory are not skipped with different bond rearrangements